Инновационный путь развития технологии создания новых лекарственных средств
Построение поверхностей потенциальной энергии (ППЭ) представляет важнейшую составную часть компьютерного эксперимента в химии, поскольку информация, содержащаяся в детальной картине этих поверхностей для молекулярной системы, поистине стоит тех серьезных затрат, которые неизбежны даже с применением мощной вычислительной техники.
Прежде всего на поверхностях потенциальной энергии находят ста
ционарные точки, то есть координаты минимумов, максимумов, седловых точек. Для того чтобы можно было говорить о существовании стабильной молекулы или молекулярного комплекса, на потенциальной поверхности основного электронного состояния должен быть минимум, энергия которого меньше энергии любой совокупности фрагментов, на которые можно разбить молекулу. Если этих минимумов несколько, то для молекулы возможно несколько изомеров. Координаты ядер, отвечающие точкам минимумов, определяют равновесные геометрические конфигурации, а энергии по отношению к соответствующим пределам диссоциации на составные части — энергии связи химической системы. Знание положений и энергий седловых точек необходимо для оценок энергий активации при рассмотрении элементарных химических реакций. Наличие минимумов с энергией выше предела диссоциации указывает на возможность образования интермедиатов в системе реагирующих молекул. Рассчитывая разности электронных энергий различных электронных состояний для тех геометрических конфигураций ядер, которые отвечают точкам минимумов, можно интерпретировать или предсказывать электронные спектры молекул.
После аппроксимации фрагментов потенциальных поверхностей в окрестностях точек минимумов можно переходить к рассмотрению движений систем ядер молекулы и прогнозировать или интерпретировать колебательно-вращательные спектры. Зная набор электронных колебательно-вращательных энергий молекулы можно с помощью формул статистической термодинамики вычислять любые термодинамические функции данного вещества. Если рассматривается молекулярная система, в которой возможно перераспределение частиц, то есть химическая реакция, то рассчитывается сечение потенциальной поверхности вдоль пути наименьшей энергии, связывающего реагенты и продукты, и затем оценивается константа скорости элементарной химической реакции. Описанная программа действий реализует схему расчетов свойств веществ без привлечения каких-либо эмпирических данных, которая представлена на рисунке 1.
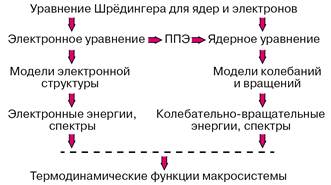
Рисунок 1 – Реализация программы расчетов свойств веществ из первых принципов
При этом результатом моделирования, которое немыслимо без компьютеров, является теоретический прогноз. Естественно, на любом промежуточном этапе этой схемы можно привлекать доступную экспериментальную информацию и вносить в компьютерное моделирование эмпирические элементы: при правильно сформулированной задаче ценность предсказаний не уменьшается, а становится более надежной. Расчеты геометрического строения и колебательных спектров молекул активно проводятся экспериментаторами, подтверждая результаты измерений квантово-химическим моделированием [4,5,6].
Молекулярная механика представляет собой совокупность методов априорного определения геометрического строения и энергии молекул на основе модели, в которой электроны системы явно не рассматриваются. Поверхность потенциальной энергии, которая в квантово-химических моделях подлежит прямому расчету, здесь аппроксимируется определенными эмпирическими функциями разной степени сложности, представляющими собой, например, суммы парных потенциалов взаимодействия атомов. Эти потенциальные функции, определяющие так называемое силовое поле молекулы, содержат некоторые параметры, численное значение которых выбирается оптимальным образом так, чтобы получить согласие рассчитанных и экспериментальных характеристик молекулы. В простейшем случае параметрами являются равновесные межъядерные расстояния и валентные углы, а также силовые постоянные, то есть коэффициенты жесткости упругих сил, связывающих пары атомов. Метод основан на допущении возможности переноса этих параметров из одной молекулы в другую, так что численные значения параметров, подобранные для некоторых простых молекул, используются далее при прогнозировании свойств других более сложных соединений.
Простейшие модели молекулярной механики учитывают растяжения связей, деформацию валентных и двугранных углов, взаимодействие валентно несвязанных атомов, называемое также Ван-дер-Ваальсовым взаимодействием, электростатические вклады и т.д.:
![]() , (1)
, (1)
где Uраст – энергия растяжения связей;
Uдеф – энергия деформацию валентных углов;
Uторс – энергия деформацию двугранных углов;
Uвдв – энергия Ван-дер-Ваальсового взаимодействия;
Uэл-стат – энергия электростатических вкладов.
Для каждого слагаемого записывается определенное аналитическое выражение и параметры соответствующих функций подгоняются по каким-либо свойствам базовых молекул. Например, для описания потенциальной функции предельных углеводородов при не очень высоких требованиях к точности расчета достаточно около десяти параметров.
Сумма всех перечисленных вкладов определяет энергию U молекулы как функцию геометрической конфигурации ядер, и для нахождения равновесной геометрической конфигурации исследуемой молекулы необходимо определить минимум U с помощью компьютерной программы поиска стационарных точек на многомерных потенциальных поверхностях. Таким образом, практические действия исследователя чаще всего сводятся только лишь к заданию стартовой геометрии и вызову программы оптимизации геометрических параметров из условия минимума энергии. На выдаче просматривается полученная структура и. если необходимо, анализируются энергия и ее составляющие.
Трудно переоценить роль молекулярной механики в современной химической практике. Поскольку все вычислительные проблемы относятся лишь к хорошо разработанным процедурам минимизации, даже на маломощных персональных компьютерах можно анализировать строение больших многоатомных молекул за разумное время. Возможность увидеть структуру молекулы на экране компьютера, рассмотреть ее с разных сторон, проверить возникающие предположения о стерических затруднениях и т.д. оказывает неоценимую помощь в работе. Молекулярная механика играет роль молекулярного конструктора: для первичной оценки строения интересующей нас молекулы зачастую проще собрать молекулу на компьютере, чем тратить время на поиск необходимой информации в справочной литературе. При расчетах молекулярной структуры на более высоком уровне методами квантовой химии полезно использовать координаты ядер молекулы, найденные с помощью молекулярной механики, в качестве начального приближения. Для многих задач, например для конформационного анализа, уровень моделирования методами молекулярной механики оказывается вполне достаточным для качественных и даже количественных заключений.
В каждом конкретном случае необходимо интересоваться, для каких классов соединений параметризована та версия программы, которую предполагается применять при моделировании свойств нового соединения. Особенно осторожно следует относиться к оценкам энергий, хотя и для геометрических конфигураций возможны грубые ошибки.[5,6,7]
Другие рефераты на тему «Химия»:
- Помутнение как характеристическое свойство оксиэтилированных ПАВ и полимеров
- Поверхностно-активные полимеры
- Лантаноиды и актиноиды
- Инструментальные методы анализа веществ
- Электрохимические методы анализа и их современное аппаратурное оформление - обзор WEB–сайтов фирм–продавцов химико-аналитического оборудования